:::
認識化學系
本系教育目標旨在培養化學專業人才與中等學校自然及化學專業師資,授課著重理論及應用性。本系所現有師資為專任教授22人,另外尚有與中央研究院合聘教授3位與2位外籍客座教授,在有機、無機、應用分析及物理化學四個學門的基礎上發展跨領域之教學研究合作計畫。本系一向秉持著教學與研究並重,研究設備亦不斷的更新。本系所的研究計畫大部分來自國科會的經費補助。此外,本系提供成績優良或清寒學生獎助學金,研究生可支領助教獎學金、研究獎學金和研究計畫所提供的博士班學生獎學金。
熱門訊息
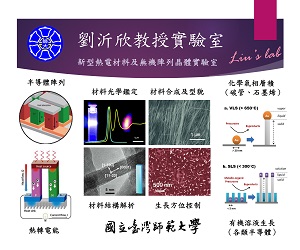
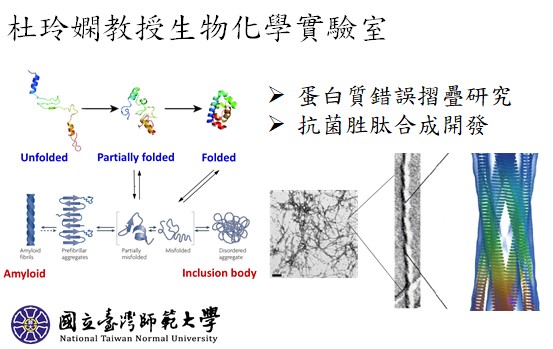
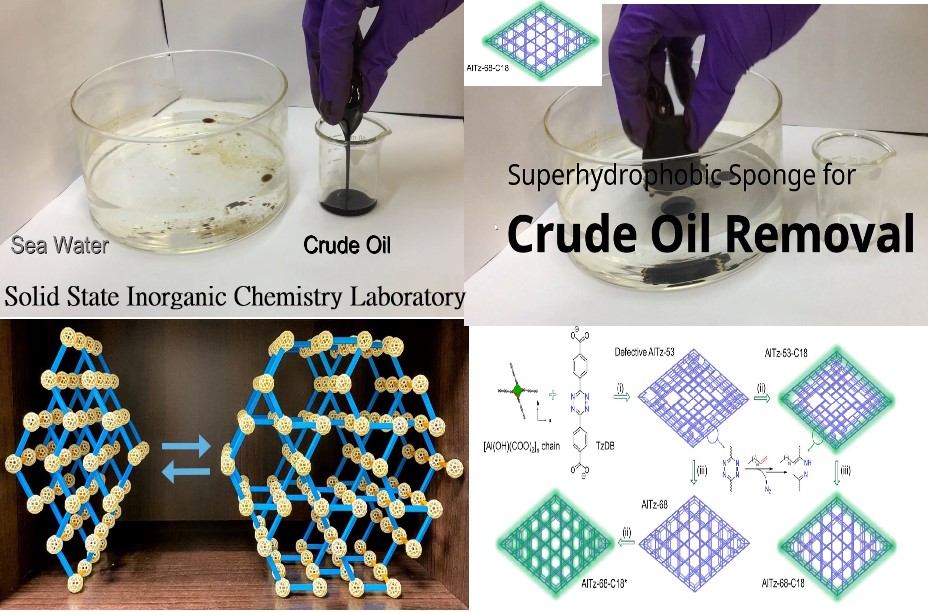
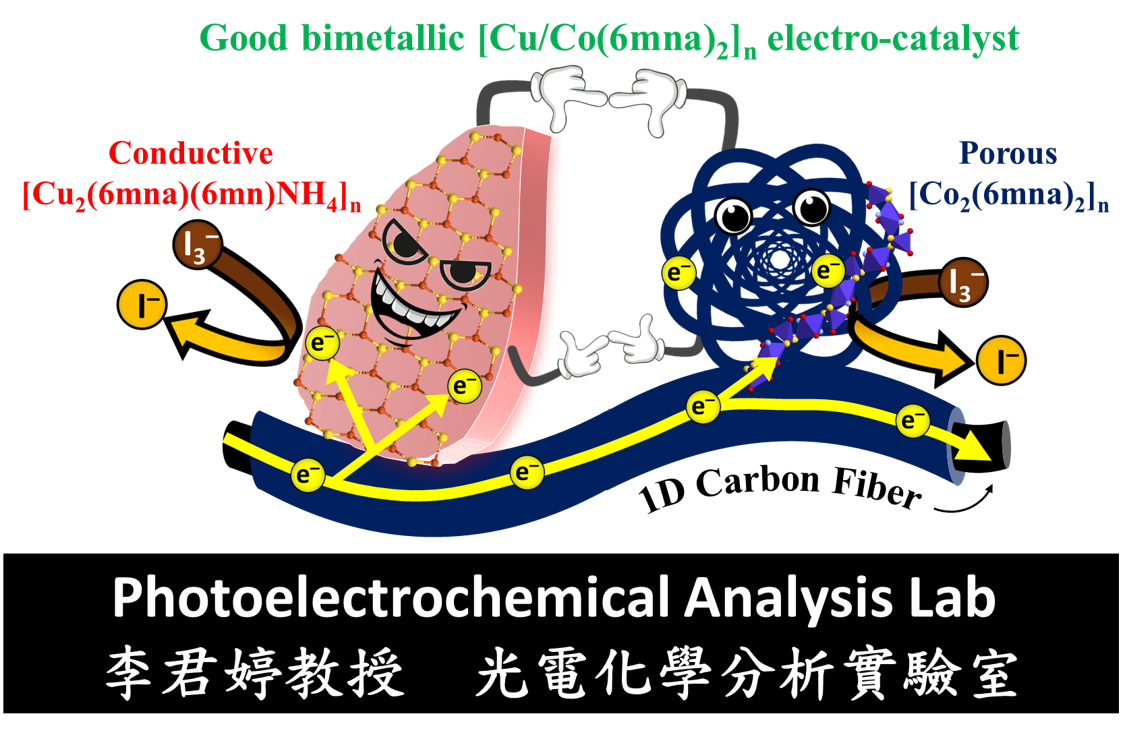
化學系研究計畫一覽表
恭賀 ! 本系陳家俊教授、林民生客座教授榮獲「全球前2%頂尖科學家榜單(World’s Top 2% Scientists 2022)」殊榮 。
恭賀 ! 本系簡敦誠教授榮獲112年教學優良獎。
恭賀 ! 本系76級系友林明裕榮任教育部政務次長。
恭賀 ! 本系張煥正教授(中研院合聘)、陳家俊教授榮獲「全球前2%頂尖科學家榜單(World’s Top 2% Scientists 2022)」殊榮 。
恭賀 ! 本系張一知教授榮獲111年度教師教學獎勵「教學傑出獎」陳家俊教授榮獲「教學優良獎」。
恭賀 ! 本系陳家俊教授榮獲「全球前2%頂尖科學家榜單(World’s Top 2% Scientists 2020)」殊榮 。
恭賀 ! 本系林文偉教授、陳頌方教授榮獲110年度教師教學獎勵教學優良獎。
恭賀 ! 李位仁教授榮獲科技部「109年度傑出研究獎」。
恭賀 ! 張鈞智博士(2016臺師大化學系)榮任文化大學化學工程系助理教授。
恭賀 ! 葉丞豪博士(2016臺師大化學系)榮任逢甲大學材料科學與工程系助理教授。
恭賀 ! 張一知教授獲選本校108學年度服務傑出教師。
恭賀 ! 李位仁教授成功研發「超氧化岐化酶(SOD)人工酵素」,技轉金額逾千萬元締造本校新猶。
恭賀 ! 葉名倉教授榮獲「中國化學會會誌」2019年最佳論文獎
恭賀!本系碩士生謝宗恩(劉沂欣教授指導)榮獲2019年中國化學會張昭鼎無機化學研究論文獎的優等獎
恭賀!本系碩士生林于寬(葉怡均教授指導)榮獲2018年中國化學會台灣神隆分析化學研究生論文獎優等獎
恭賀!本系碩士生林潔(葉怡均教授指導)榮獲2018年中國化學會台灣神隆分析化學研究生論文獎佳作獎
恭賀!孫英傑教授榮獲本校107年度「教學優良獎」
恭賀!本系碩士生倪丞緯(李以仁教授指導)榮獲2017年中國化學會物理化學研究生論文優等獎
恭賀!本系碩士生陳巧穎(李以仁教授指導)榮獲2018年生物物理年會壁報競賽第三名
恭賀!陳家俊教授榮獲財團法人傑出人才發展基金會 「傑出人才講座」
恭賀!葉名倉教授、謝明惠教授榮獲本校核聘為107年度「特聘教授」
恭賀!王禎翰教授、李祐慈教授榮獲本校核聘為107年度「優聘教授」
恭賀!吳學亮教授榮獲第19屆財團法人水木化學文教基金會 「傑出青年學者獎」
恭賀!李以仁教授研究成果榮登PNAS : Proceedings of the national academy of sciences of the United States of America
恭賀!陳焜銘教授、姚清發教授、陳家俊教授榮獲本校核聘為106年度「特聘教授」
恭賀!呂家榮教授、吳學亮教授榮獲本校核聘為106年度「優聘教授」
恭賀!呂家榮教授 率領 2017年國際化學奧林匹亞競賽團隊榮獲4金殊榮(團體第1)
恭賀!張一知教授連任國際化學奧林匹亞指導委員會主席(2017-2018)
恭賀!吳家誠教授、簡敦誠教授榮獲本校106年度「教學優良獎」
恭賀!本系與加州大學柏克萊分校化學系「柏克萊-台師大雙邊學術合作計畫」正式展開
恭賀!林震煌教授、李位仁教授、林文偉教授榮獲本校核聘為105年度「優聘教授」
恭賀!本系碩士生許嘉仁(孫英傑教授指導)榮獲2015年中國化學會研究生論文準佳作獎(新創藥物組)
恭賀!本系廖庭尉同學(陳家俊教授指導)榮獲 2015年「李遠哲院士暨夫人獎學金」
恭賀!本系系友 王素蘭教授 榮膺本校第15屆傑出校友
恭賀!本系系友 周櫻旻(蘇展政教授實驗室) 榮獲中國工程師學會高雄市分會104年度「青年工程師獎」
恭賀!第六屆海峽兩岸理論與計算化學會議優良壁報論文獎,本系共四篇獲獎,名單如下:許文綺、劉安倫(碩士班學生/孫英傑教授實驗室)、張鈞智(博士班學生/何嘉仁老師實驗室)、詹佑得、葉相均、黃羽柔(碩士班學生/蔡明剛老師實驗室)、林亮君、梁哲銘、盧恩平(專題及碩士班學生/蔡明剛老師實驗室)
恭賀!葉名倉教授、謝明惠教授、何嘉仁教授榮獲本校核聘為104年度「特聘教授」
恭賀!呂家榮副教授、吳學亮副教授榮獲與優聘教授同等級之獎勵
恭賀!本系碩士生于家齊(謝明惠教授指導)榮獲2014年中國化學會研究生壁報論文傑出獎
恭賀!姚清發教授 率領 2014年國際化學奧林匹亞競賽團隊榮獲2金2銀殊榮(團體第2)
恭賀!葉名倉教授、何嘉仁教授榮獲本校103年度「教學優良獎」
恭賀!全國大專學校運動會一般女子組網球雙打 第一名 廖攸雅
恭賀!全國大專學校運動會一般組羽球雙打 第三名 鄭至富
恭賀!陳家俊教授榮獲本校核聘為103年度「講座教授」
恭賀!陳家俊教授 榮獲 行政院國家科學委員會102年度傑出研究獎
恭賀!姚清發教授、林文偉教授榮獲本校核聘為103年度「特聘教授」
恭賀!謝明惠教授、陳焜銘教授榮獲本校核聘為103年度「優聘教師」
恭賀! 本系學士班 莊彥華 榮獲本校102學年度師資培育獎學金
恭賀! 林文偉副教授 榮獲 中國化學會102年度傑出青年化學獎
恭賀!林震煌教授研究室獲得 2013年NI圖形化系統設計(GSD)應用徵文競賽-進階研究組-冠軍
恭賀!本系學士班李盈萱(呂家榮副教授指導)榮獲 2013年「李遠哲院士暨夫人獎學金」
恭賀!林文偉副教授 榮獲 行政院國家科學委員會102年度吳大猷先生紀念獎
恭賀!張一知副教授 率領 2013年國際化學奧林匹亞競賽團隊榮獲3金1銀殊榮(團體第1)
恭賀!張一知副教授、李位仁副教授榮獲本校102年度「教學優良獎」
恭賀!陳家俊教授榮獲本校核聘為「特聘教授」
恭賀!葉名倉教授、姚清發教授、林震煌教授榮獲本校核聘為「優聘教師」
恭賀!林文偉副教授榮獲與優聘教師同等級之獎勵
恭賀!本系碩士生程健(簡敦誠教授指導)獲2012年中國化學會研究生論文獎有機化學組佳作
恭賀!本系碩士生吳冠緯(孫英傑教授指導)獲2012年中國化學會研究生論文獎新創藥物組入選
恭賀!本系65級校友王素蘭老師榮獲第16屆國家講座主持人
恭賀!本系43級校友蕭曉萍博士榮獲美國太空總署2012年度卓越科技貢獻獎
恭賀!師大化學87級畢業系友紀雅惠當選中華民國第五十屆 「十大傑出青年」
恭賀!姚清發主任 率領 2012年國際化學奧林匹亞競賽團隊榮獲3金1銀殊榮(團體第2)
恭賀!國科會召開記者會(林震煌教授發表研究成果)
恭賀!國科會科技大觀園專題報導 林震煌 教授
恭賀!方泰山教授榮獲教育部「三等教育文化專業獎章」
恭賀!何嘉仁教授榮獲本校核聘為「特聘教授」
恭賀!陳焜銘教授榮獲本校核聘為「優聘教師」
恭賀!陳家俊教授 指導 博士生周尚威 榮獲2010年「第七屆永信李天德醫藥科技獎」
恭賀!張一知副教授 高票當選2012-2013年國際化學奧林匹亞競賽「國際指導委員」
恭賀!方泰山教授 率領 2011年國際化學奧林匹亞競賽團隊榮獲3銀1銅佳績
恭賀!吳家誠教授 榮獲中華民國環境分析協會頒發「環檢領航」獎座
恭賀!本系系友 李光華教授 榮膺本校第11屆傑出校友
恭賀!謝明惠教授榮獲本校99學年度第2學期「特聘教授」
恭賀!本系系友 呂光烈教授 榮獲99年度國科會「傑出研究獎」
恭賀!陳家俊教授發表雙功能分子標靶奈米顯影劑並研製成功,榮登美國化學學會會誌(Journal of the American Chemical Society) 封面
恭賀! 臺灣師範大學化學系進榜世界之論文被引次數前1%